The myeloid cancers encompass myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML), affect ~10 per 100,000 individuals per year and remain lethal to the majority. Recent studies by us and others have revealed that individuals en route to developing these cancers can be identified years in advance (Abelson et al, Nature 2018 and Gu et al, Nat Genet 2022). This has raised hope that timely identification of at-risk individuals may enable early interventions to prevent progression to malignancy. Jointly funded by the Leukemia & Lymphoma Society and Blood Cancer UK, this Specialized Center of Research (SCOR) project aims to make myeloid cancer prevention a clinical reality.
Specialized Center of Research
Development of a Clinical Program for Myeloid Cancer Prevention
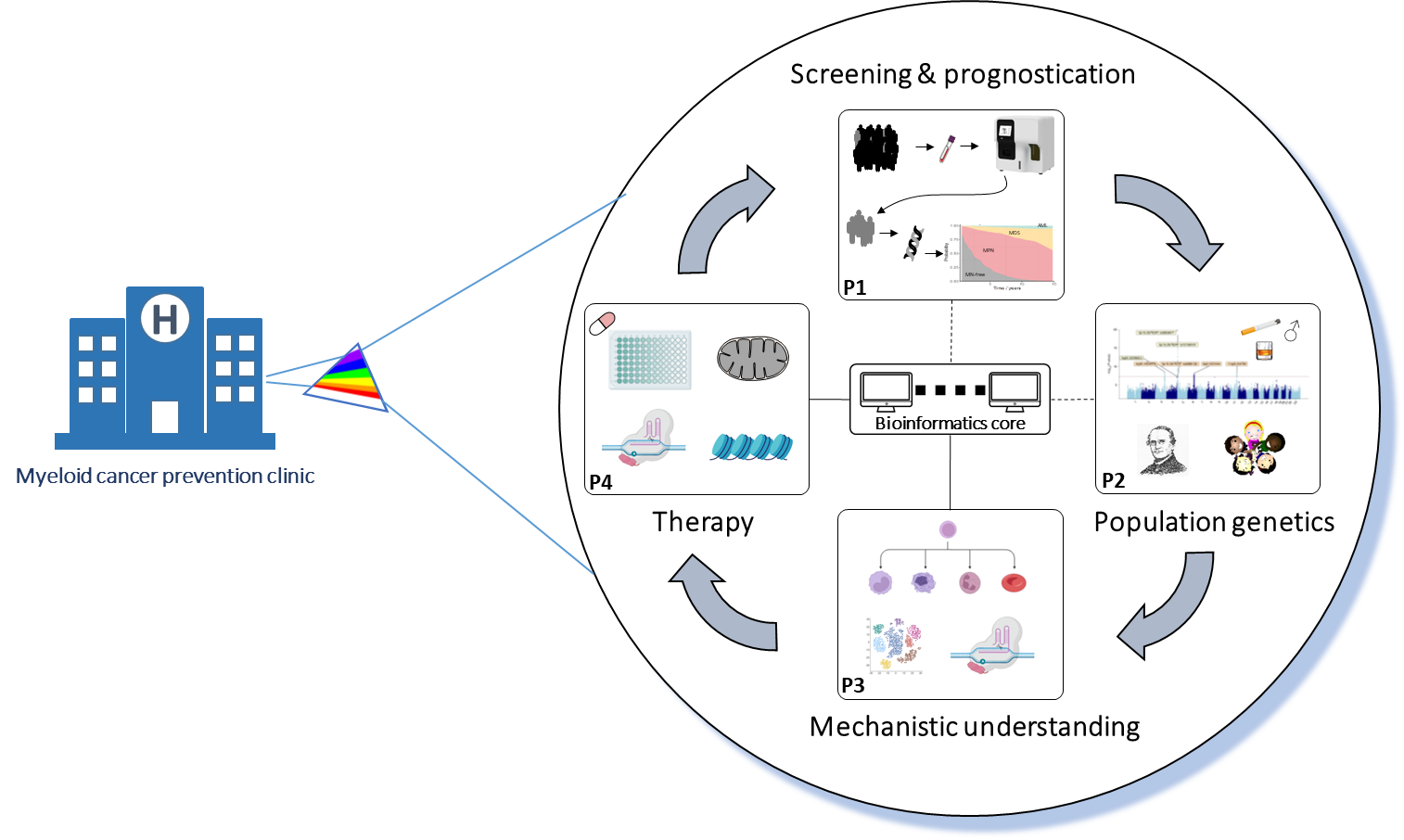
Led by Professor George Vassiliou, our team is composed of four Hematologists/Clinician Scientists (Vassiliou, Huntly, Nangalia, Green) and four Basic Scientists (Gottgens, Mendez-Ferrer, Laurenti, Kar) based at the University of Cambridge and the Wellcome Sanger Institute. Each is an international leader in myeloid cancers, clonal haematopoiesis and/or cancer early detection.
The work will be conducted under four interacting projects:
(1) Screening & Prognostication


George Vassiliou Jyoti Nangalia
Aim 1: Develop a scalable screening tool for high risk CH or CCUS
Currently most patients with CH or CCUS are identified by chance and there are no programs or screening platforms to identify them systematically. Panel gene testing could be used in this context, but it remains expensive, difficult to communicate to the public and in competition with a number of different primary care screening tests. Here, we propose to develop a machine-learning (ML) approach to analyze results of the complete blood count (CBC), a routine and inexpensive blood test, to identify individuals that may harbor high risk CH/CCUS. We will work to maximize performance by incorporating hematology analyzer raw data, integrate inherited genetics and decipher the significance of longitudinal changes in CBC parameters. Plasma proteome data from 50,000 UKB participants will also be investigated in this context. In addition, we will test the ability of our recently developed automated cytomorphology platform, Haemorasis, to discern morphological features associated with different forms of CH.
Aim 2: Develop a prognostic platform to quantify individuals’ risk of developing different myeloid cancers.
The clinical implementation of myeloid cancer prevention will rely heavily on accurate estimation of myeloid cancer risk. We have previously shown that a predictive model based on somatic mutations can be used to quantify the AML risk, however this has not been performed adequately for MDS or MPN. Also, the utility of non-genetic predictive parameters, such as blood test results, has not been investigated. Here, we will use data from large databases and from clinical patient cohorts to build a robust prognostic model that can predict the risk of AML, MDS and MPN. We have already developed a prototype model/tool, MN-Predict.
(2) Population Genetics

Aim 1: Identify new germline susceptibility loci and develop polygenic risk scores for clonal hematopoiesis (CH).
This will involve increasing the sample size of genome-wide association studies (GWAS) of CH via higher sensitivity phenotyping to increase the number of individuals with CH detectable and the use larger populationbased cohorts with linked blood-based genomic and clinical data. We will also perform cross-trait genome-wide association meta-analyses that leverage shared genetic associations between CH and closely related traits such as leukocyte telomere length, as well as a range of blood cell counts/indices, to improve the power to detect inherited variants associated with CH risk. Finally, we will leverage these cross-trait data to develop polygenic risk scores as germline predictors of CH and subsequent progression to hematological malignancies.
Aim 2: Identify the candidate causal variants and their target genes at known and newly discovered CH risk loci.
This will involve statistical fine mapping of germline genetic associations in each region known or newly identified to be associated with CH risk to define the most likely causal variants that confer CH risk. The genes through which these variants are likely to impact on CH risk will be shortlisted via integration of bulk gene expression quantitative trait loci (eQTLs) and single-cell RNA- /ATAC-Seq data to derive cell type-specific functional genomic annotations with the GWAS data. The effect of each causal variant on mutation acquisition versus clonal expansion will also be evaluated with large-scale clonal evolution modeling from single timepoint samples.
Aim 3: Identify new and potentially modifiable causes of CH and its progression to myeloid neoplasia (MN).
This will involve state-of-the-art causal inference modeling using Mendelian randomization (MR) tapping into largescale exposure/risk factor genetic association datasets, including for circulating proteomic and metabolomic exposures on >50,000 UK Biobank (UKB) participants. We will use these data to implicate novel causes of CH and find causal consequences via which CH may confer blood cancer risk, improving our mechanistic un
(3) Mechanistic Understanding


Bertie Göttgens Elisa Laurenti
Aim:to deliver mechanistic understanding of how specific germline and somatic loci associated with CH impact HSC function.
Project 3 will assess the functional relevance of somatic mutations/germline loci identified in Project 2, identifying new facets of HSC biology and providing the mechanistic insights essential to develop new therapeutics interventions (Project 4). We seek to quantify the impact of loci/variants driving clonal expansion on HSC cellular functions and Identify targetable molecular mechanisms by which specific CH-associated loci/variants modify HSC function.
(4) Therapy


Simón Méndez-Ferrer Brian Huntly
Aim (1): Functional genomic identification and validation of CH vulnerabilities ex vivo and in vivo.
Aim (2): Development of in vivo assays to test therapeutic targeting of CH mechanisms.
Aim (3): Validation and pre-clinical testing of novel CH mechanistic targets.
We hypothesize that the mutations and other changes that drive CH will confer therapeutic vulnerabilities to CH clones, either shared across mutations or specific to individual mutations. These, can then be therapeutically targeted to delay MN development or even prevent this altogether.